浅述周环反应及相关理论
注:还完全没写完捏,慎看()
周环反应 Pericyclic Reaction
周环反应概述
历史背景
在 20 世纪 60 年代之前,人们认为有机中的反应性是由以下因素主导的:
- 反应物和产物的相对稳定性(即热力学控制);
- 如反应物或产物的芳香性。
- 过渡态的早期/晚期性质(Hammond 假设,动力学控制);
- 几何效应,如张力、空间相互作用、氢键、邻基效应(熵效应);
- 静电效应,如官能团(如羰基)的极性。
在 20 世纪 60 年代早期维生素 B12 的合成过程中,罗伯特·伍德沃德 (Robert Woodward) 总结道(了解完整故事参见 Chem. Soc. Special Publications (Aromaticity), 1967, 21, 217),上述因素都不能合理解释以下反应。当时人们认为,使用简单的分子模型(另一位伟大的有机化学家和诺贝尔奖得主德里克·巴顿 (Derek Barton) 于 1951 年开创了定量模型),预测产物为 J,因为生成 H 的路径在空间上更加严苛,因此形成的可能性较小。然而事实情况恰恰相反: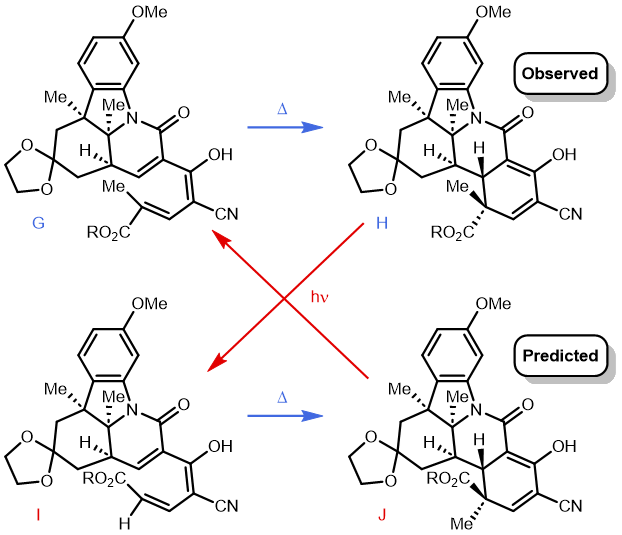
一种新的解释基于立体电子效应,即电子的三维性质和它们的相位关系可以在上述各种因素中占优势。这种周环反应的立体电子控制理论是伍德沃德与理论学家霍夫曼 (Roald Hoffmann) 一起使用分子轨道对称性守恒(在后文会详细讲解)推导出来的。伍德沃德在霍夫曼(和福井 Fukui)因这项工作(发表于 J. Am. Chem. Soc., 1965, 87, 395,这篇文章的评论在这里)获诺贝尔奖之前就去世了。立体电子效应(更准确地说是分子波函数的三维性质)对解释反应性至关重要,这对后人理解有机化学产生了巨大的影响。
周环反应的定义与性质
周环反应是指化学键在协同的环状过渡状态下形成或断裂的反应。协同反应 (Concerted Reaction) 是指在反应过程中不涉及中间产物的反应(左图)。图中还显示了具有离散中间体的分步非协同、非周环反应(右图)。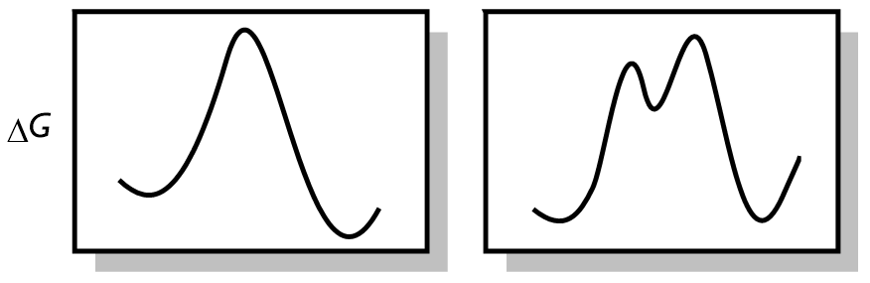
因此,理解周环反应必然需要理解如何控制它们的过渡态。
周环反应通常具有这些特征性质(尽管通常不难发现这些规则的例外情况):
- 没有亲核或亲电组分。这意味着在写机理“推箭头”时,箭头没有起点也没有终点,可以按顺时针或逆时针方向进行推箭。
- 通常情况下,周环反应不需要任何催化剂来促进。然而,许多过渡金属配合物可以通过 d 轨道参与催化周环反应。Lewis 酸还可以直接催化多种形式的周环反应,或者通过改变反应的机理,使其成为一个分步的过程,不再是真正的周环反应。
- 周环反应通常表现出非常高的立体选择性。
- 周环反应通常可以用光(在正文中表示为hν)和热(在正文中表示为 Δ)来促进。通常,两组条件下反应的立体化学是不同的(最初人们认为它们总是相反的)。目前对光化学路线的思考更为复杂。
- 周环反应可以在没有溶剂的气相中发生。溶剂对反应速率的影响相对较小(除非反应物本身恰好带电荷)。人们曾对用水加速周环反应(大约 10 到 100 倍)进行了大量研究,但这种加速很大程度上是由于过渡态中氢键的形成。
- 可以使用字面意义上的机械力(如研磨)来影响周环反应结果(机械力化学 Mechanochemistry)。
- 对于那些体积大幅减少的反应,可以通过加压来加速反应。还可以通过设计催化剂(“分子容器”),提供合适的“空腔”以促进周环反应,如环加成反应。
- 与亲核/亲电反应不同,周环反应是不寻常的,因为已知的催化它们的酶少得惊人。人工催化抗体(‘abzyme’)可以被创造出来执行这一壮举,‘Diels-Alderase’可以对环加成反应起作用,Chorismate mutase 可以催化 Claisen 重排,其通过上述第 5 和第 7 点发挥催化作用。
有机周环反应的主要类别
在这里,我们没有添加也可以描述为周环的丰富多样的无机反应。
有机周环反应有四个主要的类别,涵盖了非常广泛的有机和有机金属化学,并为理解合成和有机化学的微妙方面奠定了丰富的基础。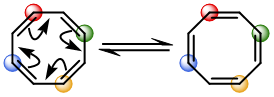
主要类别
在处理真正的周环反应之前,我们应该排除键迁移异构化(如下所示,一个相对较慢的过程,ms)或共振(如在苯中,一个极快的过程,fs)。对于周环反应来说,必须有至少一个 σ 键形成或断裂。
电环化反应
电环化反应涉及在线性共轭 π 体系的两端间协同形成一个 σ 键,或者一个 σ 键断裂产生线性共轭体系的逆反应。这里的“共轭”是指由 C-C 多重键或孤对电子体系。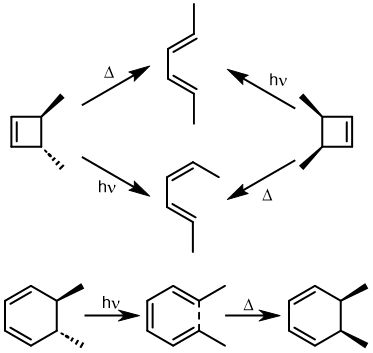

环加成和环消除反应
环加成反应是指在两个或多个共轭 π 体系的末端间协同形成两个或多个 σ 键。逆反应涉及两个或更多 σ 键的协同断裂,以产生两个或多个共轭 π 键。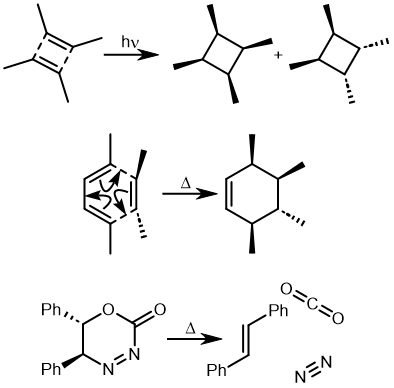
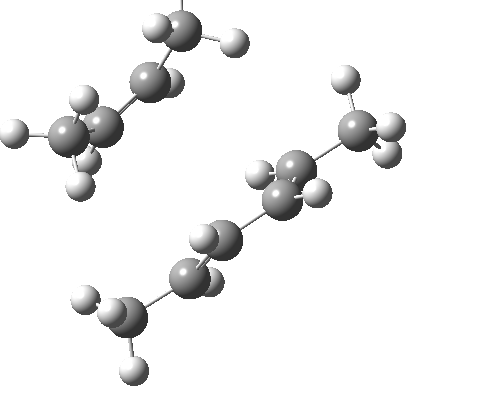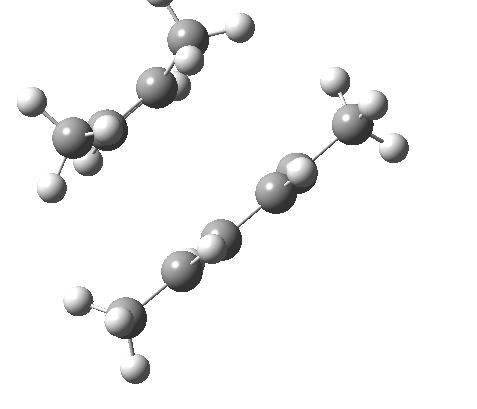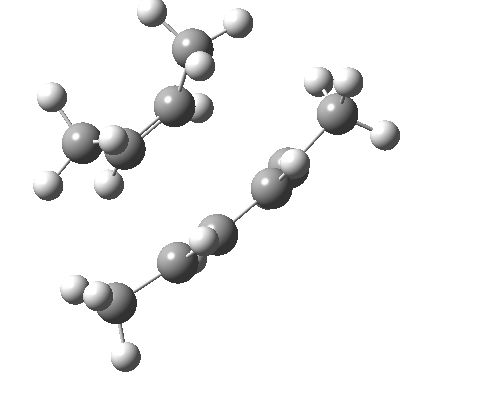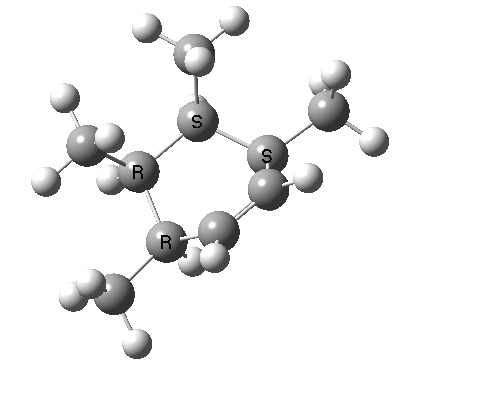

上图最后一个例子是一个三组分的 $_π2_s+ _π2_s+ _π2_s$ 环消除反应。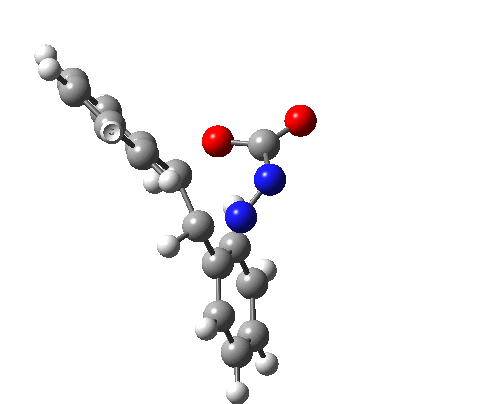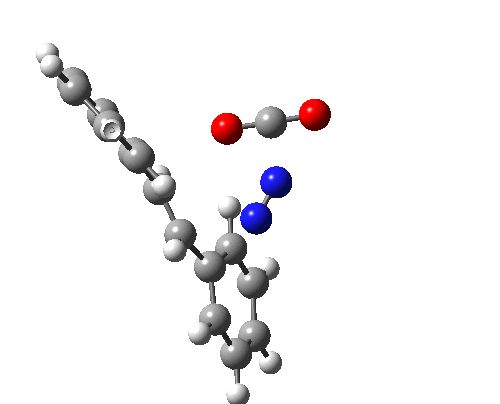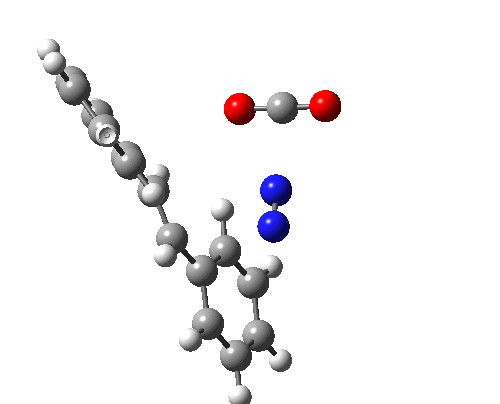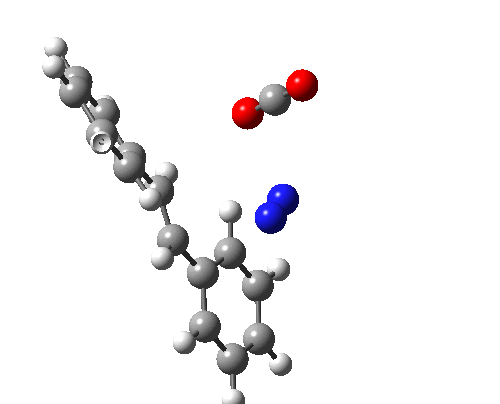
而对于两个 σ 键的形成或裂解发生在单个原子上的特定反应,被称为螯键反应: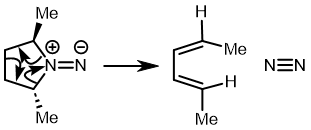
sigma 迁移反应
sigma 迁移反应涉及原子或基团从一个连接点到共轭体系再到另一个连接点的协同迁移,在此过程中,一个 σ 键断裂,并形成一个 σ 键。sigma 迁移可以根据迁移基团的长度和沿主链迁移的长度进行分类:

Ene 反应
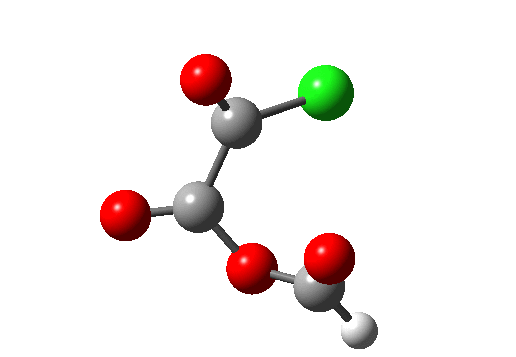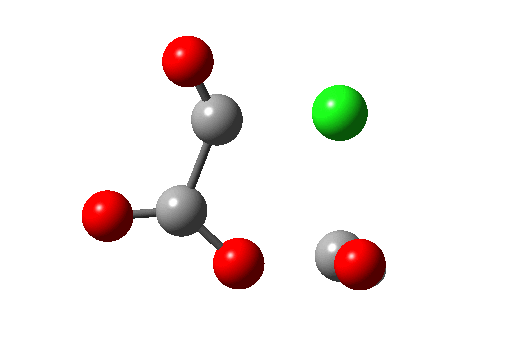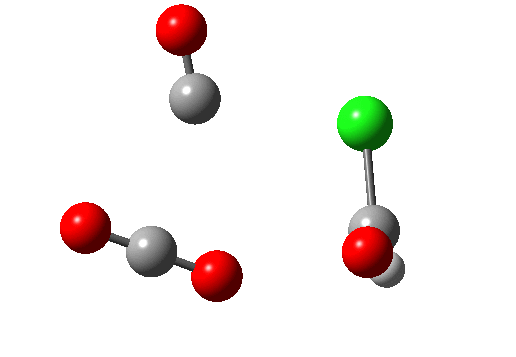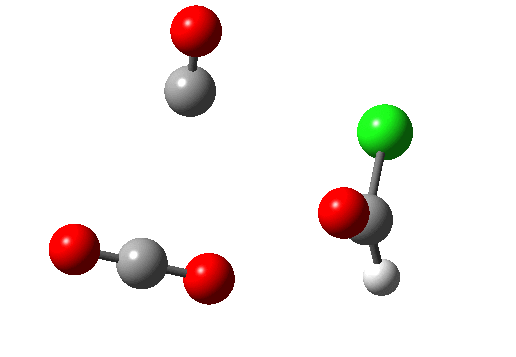
Ene 反应涉及在协同的环状过渡态中数量不等的 σ 键的形成和断裂。下图右上的例子(红色)显示了使用草酰氯将羧酸转化为酰氯的部分机理(三个键断裂,一个键生成)。该反应还具有络合消除的特点。右下角蓝色的例子 (DOI:10.1038/Nature07010),涉及羟卡宾的制备,此羟卡宾可通过氘的同位素效应抑制隧穿效应以显著增加稳定性,该效应被认为是导致未氘代物种寿命极短的原因(未氘代羟卡宾在 11K 时寿命为 2 小时,而氘代者为 2000 年,同位素效应约为 900 万)。所有这些例子都是 6 电子/4N+2 的热过程。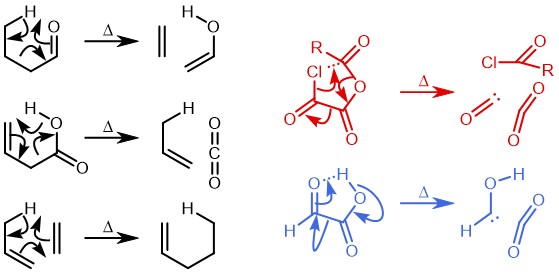

次要类别
Multiple personality pericyclics.
一种是所谓的双周环反应,它同时有着两个或两个以上独立的周环反应的特征。最初被报道的是环戊二烯二聚的机理,现在认为这种机理比预想更普遍。它同时具有$_π2_s+_π4_s$和$_π4_s+_π2_s$环加成的特征(它们是不一样的)以及[3,3] sigma 迁移。
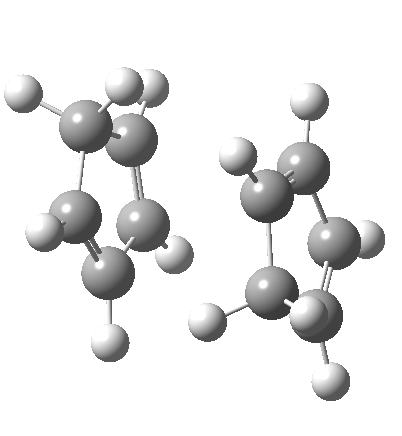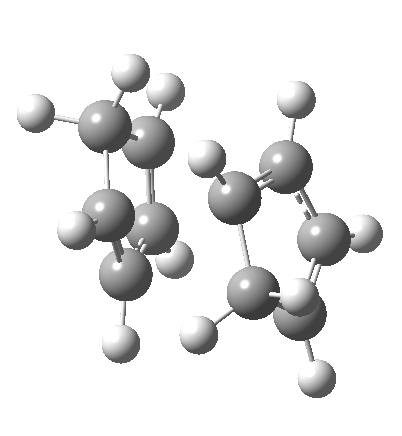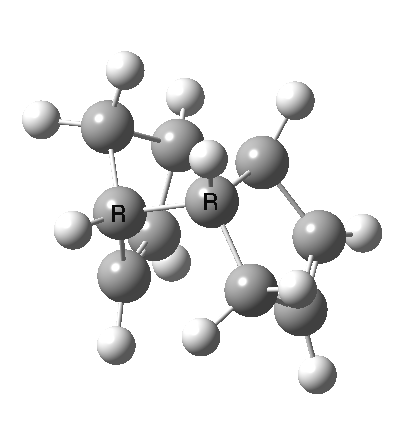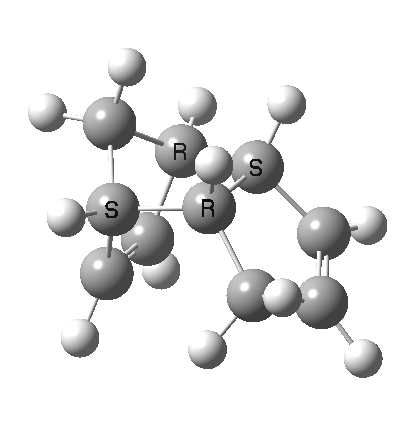
另一种具有多重性质的周环反应类型涉及环丙烷环,它通常被认为是名义上的烯烃。这应该被视为更接近于三烯的电环化反应(如上所述,环丙烷充当烯烃)还是[1,5] sigma 迁移反应(环丙烷充当单键)?这也可以看作是同芳香过渡态的表现(见后文)。
理论解释
故事始于埃瓦里斯特·伽罗瓦 (Évariste Galois, 1811-1832),他建立了基于排列对称性的理论,即我们现在所知的群论。伍德沃德和霍夫曼最初基于最高占据分子轨道的性质的原始解释是由 Longuet-Higgins 和 Abrahamson 在伽罗瓦群论的帮助下形成的 (DOI:10.1021/ja01087a033,另见10.1021/ja01087a033)。这包括为所考虑的反应生成所谓的轨道能级相关图,然后以反应物和产物轨道的群论对称性完全匹配的方式进行反应。这种方法虽然在理论上严格,但并不容易适用于大多数没有形式对称性的更复杂的反应。后文概述了避免这一问题的两种简单得多的方法,第一个基于过渡态的芳香性,第二个基于前线轨道。
分子轨道对称守恒原理(Longuet-Higgins 和 Abrahamson)
让我们首先定义 1s 和 2p 轨道相对于对称面或对称轴的对称性,如下所示:
通过考虑由两个或多个原子轨道重叠形成的分子轨道的对称性,我们可以更进一步得到:
- 由两个 s 轨道重叠形成的 MO(σ 键):
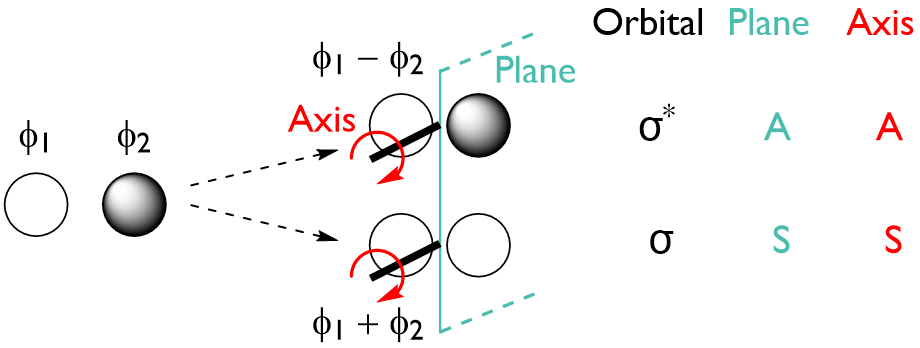
- 由两个 p 轨道重叠形成的 MO(σ 键):
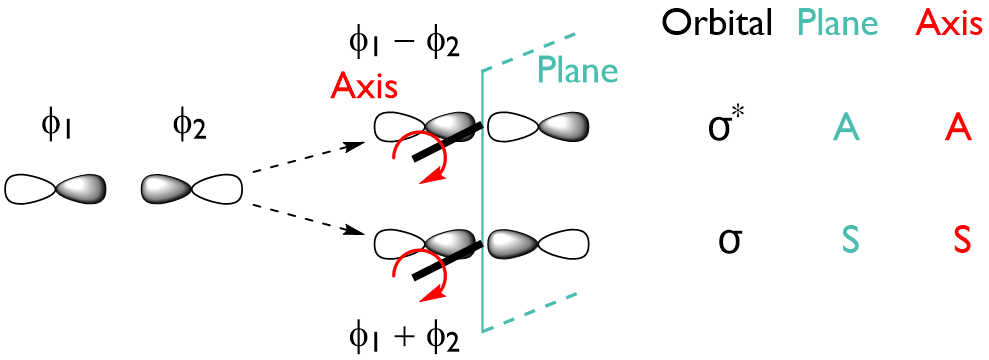
- 由两个 p 轨道平行重叠形成的 MO(π 键):
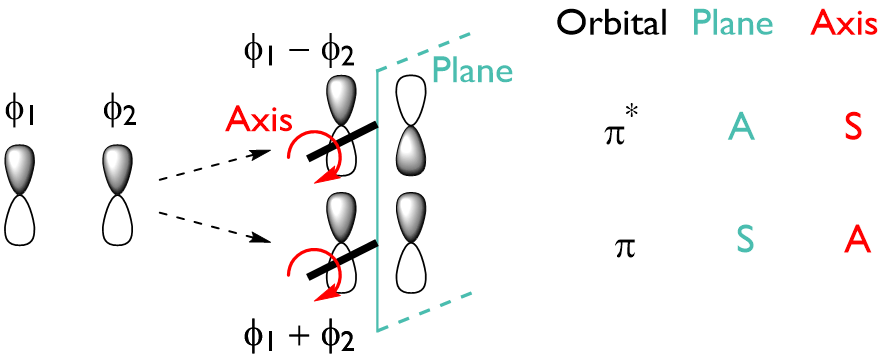
我们现在可以使用这些基本轨道来构建两个相互转化的分子环丁烯和丁二烯的相关分子轨道,目的是跟踪这两组轨道在一个分子转化为另一个分子时会如何变化。要特别注意的是,我们只需要构建明确参与反应的 MO;大多数 σ 框架保持不变,不需要考虑由此派生的轨道: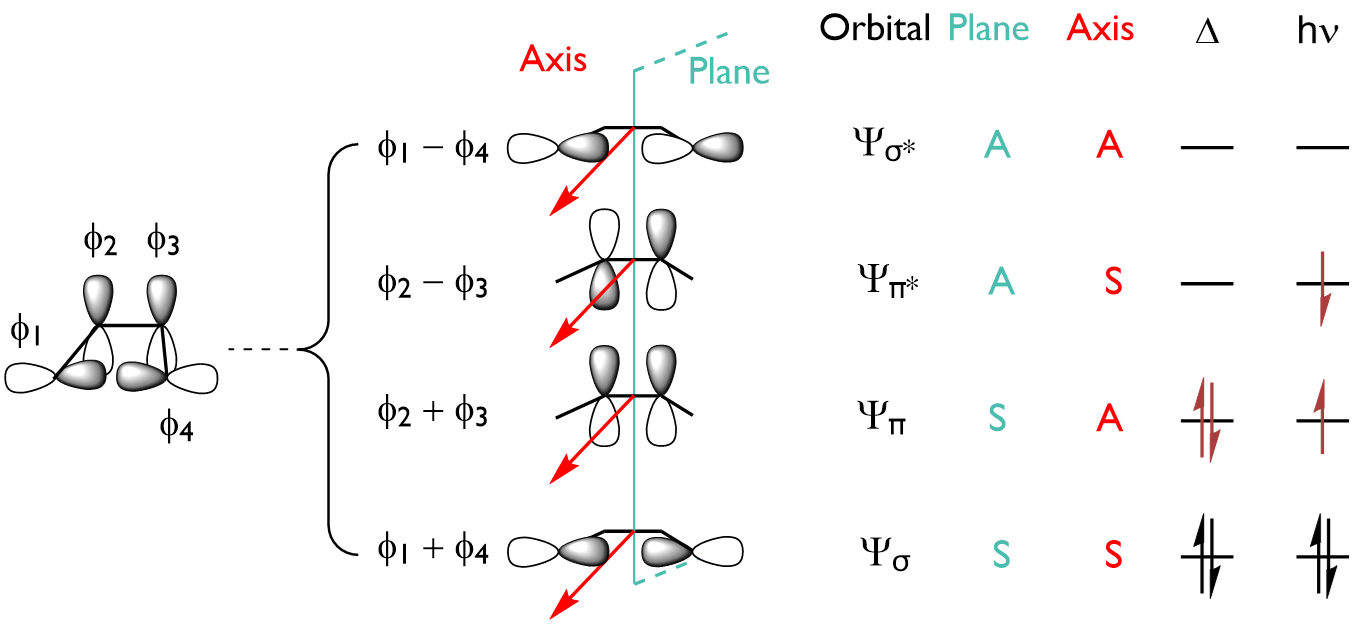
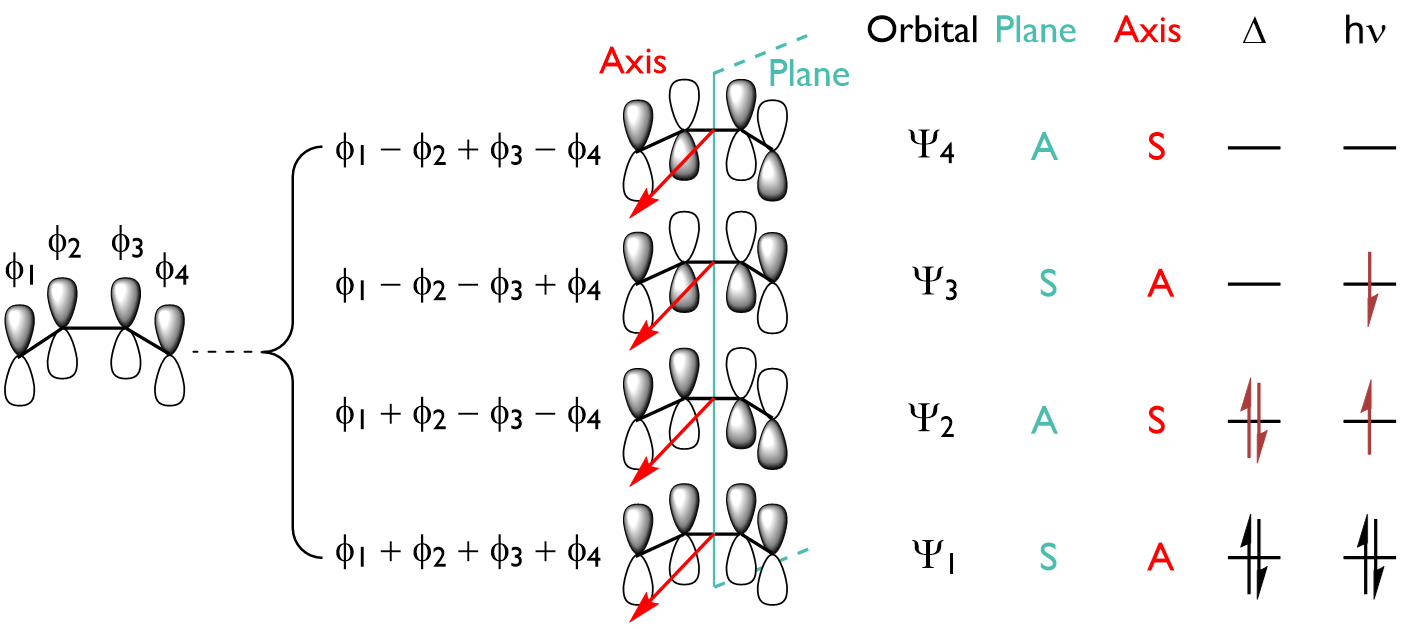
为了使环丁烯和丁二烯相互转化,标记为 $\psi_1$, $\psi_2$, $\psi_3$, $\psi_4$ 的四个 MO 必须转化为 $\psi_\sigma$, $\psi_\pi$, $\psi_{\pi^*}$, $\psi_{\sigma^*}$。有两种立体化学上截然不同的方法可以实现这一点:
Conrotation 顺旋:
Disrotation 对旋: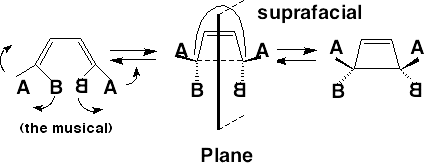
这时可以根据以下规则构建反应的能级相关图:在反应过程中,相同对称性的两个轨道不能交叉,而不同对称性的轨道可以交叉。最优势的途径是产生与反应物(绿色)相同的电子激发产物的途径。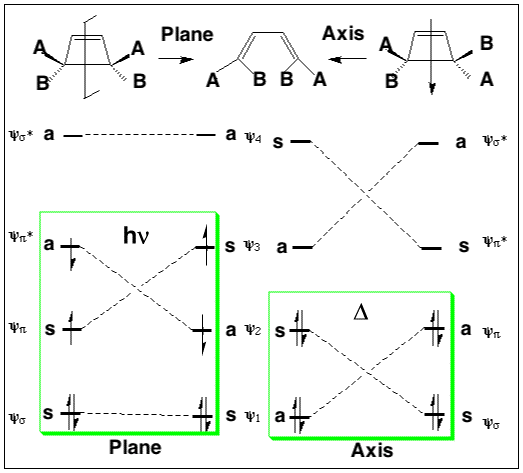
导致产物形成的电子态高于反应物的途径被称为是“禁阻”的(红色)。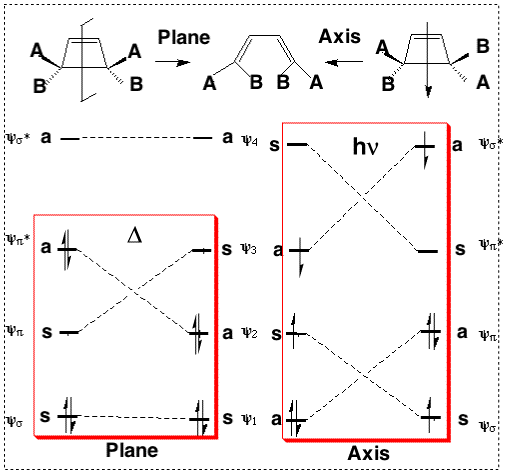
虽然对于基态通常可以很好地遵循此规则,但当“允许”路径中空间或几何张力促进“禁阻”路径时,它可能会被推翻。而光化学反应的情况实际上更为复杂,许多证据表明反应并不总遵守伍德沃德-霍夫曼规则。这些能级相关图可以推广到任何具有适当对称性的电环反应。然而,能级相关图不太容易应用于没有对称性的反应,我们需要一种更能容忍对称性破坏的方法。在下一节中,我们将介绍这种方法。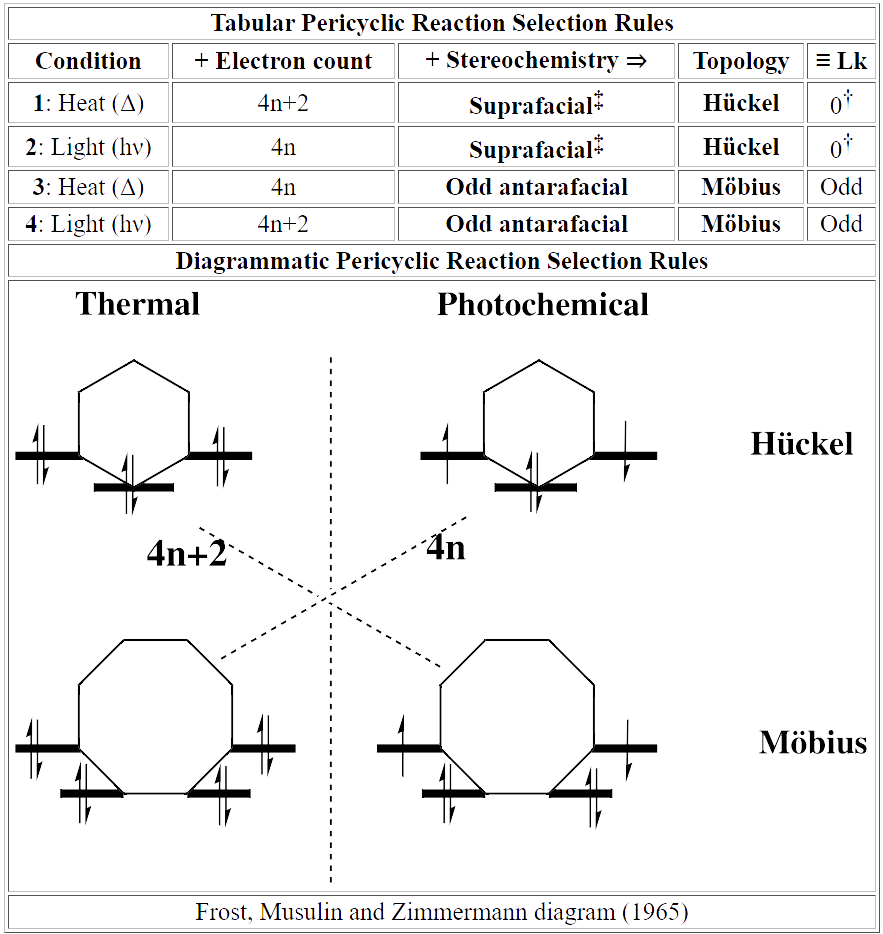
基于过渡态芳香性的选律(Dewar 和 Zimmermann)
Dewar 和 Zimmerman 独立研究发现,周环反应的分子轨道能级相关图的对称性与使用 Hückel 理论获得的芳香族分子非常相似。例如,己三烯电环化为环己二烯的分子轨道图在其过渡态与苯的基态分子轨道非常相似;
这些规则的前提是有利的周环反应将通过芳香过渡态进行。事实上,存在两种基本的相关芳香性,它们在对称性/拓扑性质上有根本的不同。根据反应是由热还是光 (Δ 或 hν) 促进的,我们可以将规则分为五类。
热(闭壳层电子组态),无波相转变(具有对称面/同面):Hückel 芳香,Lk=0。
己三烯的同面轨道相关图可以参照典型的芳香族分子苯来概括。苯分子具有基于对称面的同面 (suprafacial) π 共轭。这意味着苯中的 π 电子密度沿着分子的顶面或底面循环连续,并在将苯一分为二的对称平面上对称地反映出来。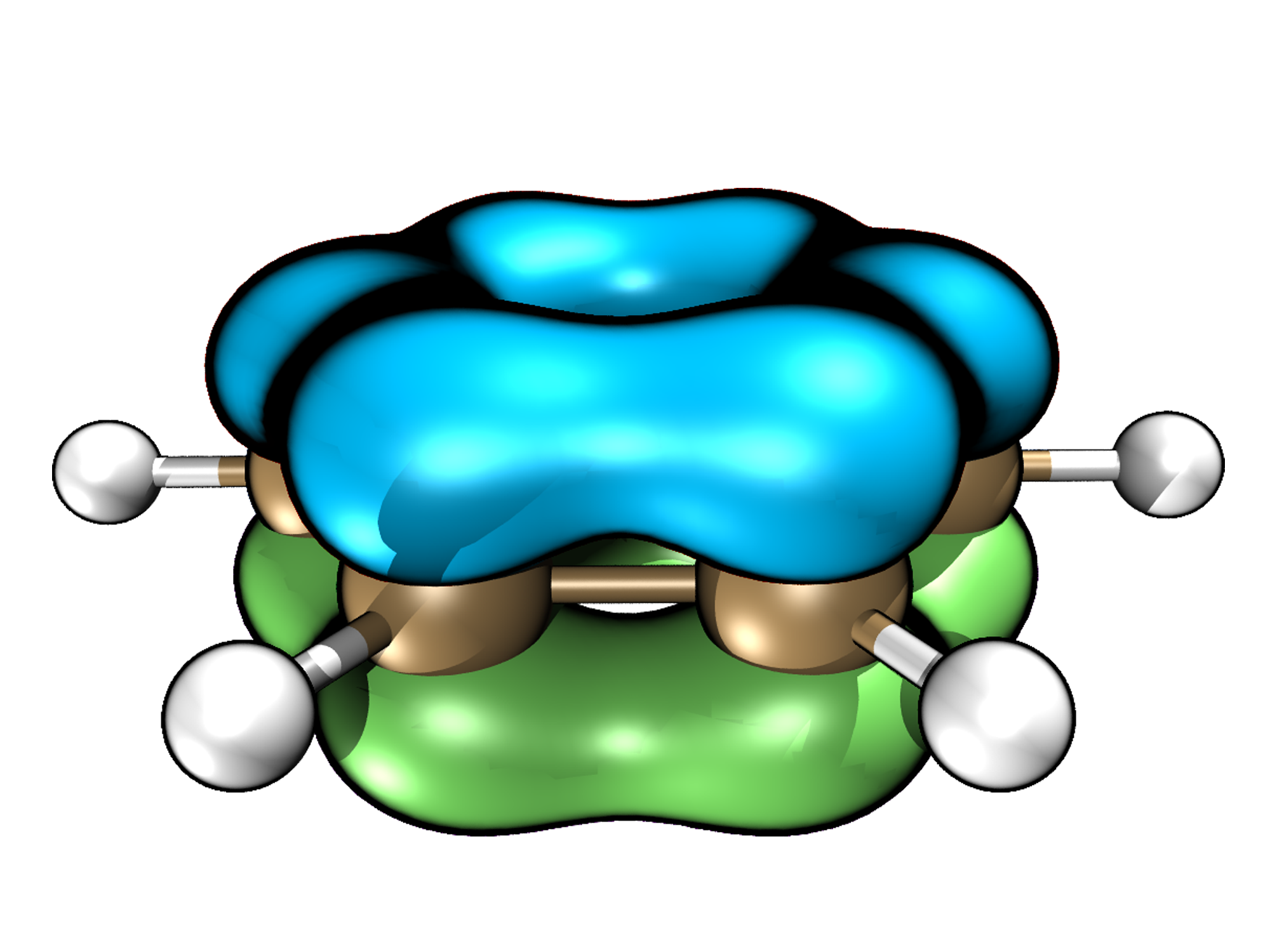
在数学中,这种类型的拓扑对象更普遍地称为二元链环,其环绕数 Lk 等于零。
苯特别稳定(芳香 aromatic),因为循环共轭的 π 电子双占据充满成键分子轨道,其总 π 电子数符合公式 4n+2:Hückel 规则,其中 n = 0, 1, 2 等(不过这个规则实际上不是由 Hückel 制定的,而是由有机化学家威廉·冯·艾格斯·多林制定的,参见 DOI: 10.1021/ ja01146a537)。
这种方法的优点是芳香性对对称性的扰动相当稳定(例如环芳烃中的苯环可以弯曲高达 30°,并仍然保持其芳香性,可用 NMR 证明)。
因此我们得出结论:如果电子数对应于 4n+2,则闭合壳层周环反应将全部通过 Hückel 立体化学同面进行。因此,这条规则现在可以应用于不对称的周环反应,这当然是迄今为止最常见的。
光(开壳层电子组态),无波相转变(具有对称面/同面):Hückel 芳香,Lk=0。
在 Hückel 之后的一段时间,人们发现,通过(光化学)将一个电子从双占据轨道激发到更高能量的分子轨道以产生所谓的开壳层分子,只有当 π 电子数为 4n 而非 4n+2 时,同面共轭 π 体系的激发态(三重态)才与芳香性相关。这种开壳层芳香族分子的一个例子是环丁二烯的三重态。
应用与之前相同的类比,我们推断如果电子数对应于4n,则开壳层周环反应(通常意味着光化学激活)将通过 Hückel 立体化学在同面发生。更现代的方法不使用过渡态来推断光化学反应的立体化学,而是使用势能面的另一个特征,即圆锥交错点(conical intersection,即 CI,俗称漏斗),通常可以得到相同的立体化学结论。
热(闭壳层电子组态),一次波相转变(具有对称轴/一个异面):Möbius 芳香,Lk=1。
与轨道能级相关图相关的异面立体化学模式类似于 Möbius 拓扑,August Ferdinand Möbius 发明的著名条带(另见 Johann Benedict Listing)。将环状烯烃“条”的 π 电子体系进行 180°(半)扭转(这样就在分子中形成了一个 C2 对称轴),就可以形成反共轭模式。Edgar Heilbronner 在 1964 年从理论上推测(当时还不知道这样的分子),如果这种扭转共轭体系包含 4n 个共轭 π 电子并占据 2n 个 MO,那么它就是一个闭壳层分子。后来证明,它也是芳香的。从这些 4n 电子导出的电子密度的拓扑分析表明 (DOI: 10.1039/b810301a) 它的拓扑对象称为一元链环(纽结),连接数 Lk 为 1 (这种结一般称为平凡纽结(unknot))。正如对只有 C2 对称轴的物体所预期的那样,它具有 C2 对称的手性(disymmetric chirality)。

Möbius 分子的第一个明确例子是在 1998 年提出的阳离子 $\mathrm{C_9H^+_9}$ (它有 8 个共轭 π 电子)。它是一个 4n 体系,其中 n=2,其 $\mathrm{^1H\space NMR}$ 表明它确实是芳香的,由此推断它是 Möbius 芳香体系。
2003年,首次制成 Möbius 环烯晶体 (DOI: 10.1021/cr030092l),对称轴终于被揭示出来。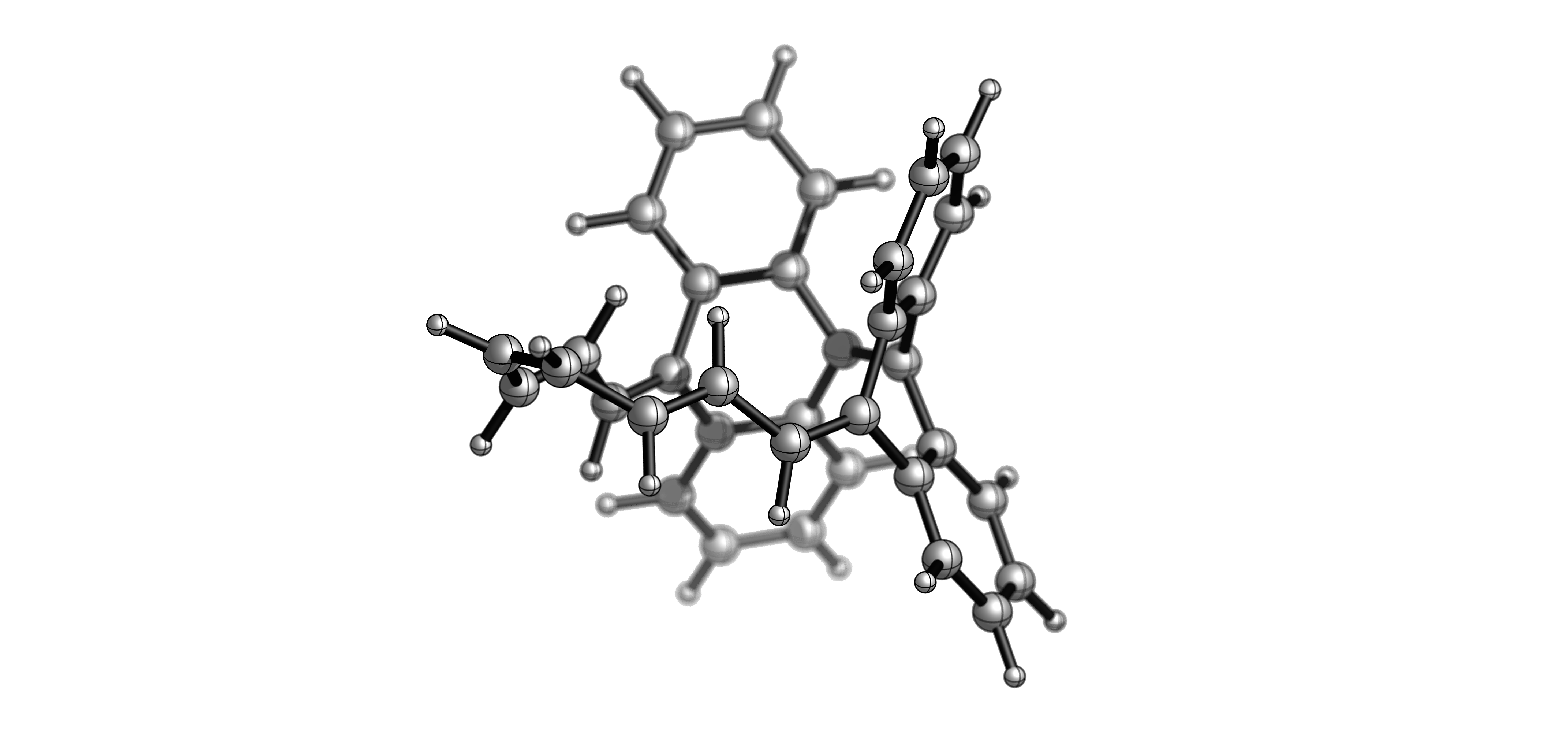
自 2006 年起,已经发现了相当多的 Möbius 芳香体系。
因此,使用与之前相同的类比,如果电子数对应于 4n,热周环反应将通过 Möbius 芳香过渡态异面进行。
光(开壳层电子组态),一次波相转变(具有对称轴/一个异面):Möbius 芳香,Lk=1。
正如光化学(激发态)Hückel 系统符合 4n 电子规则,光化学 Möbius 系统符合 4n+2 个电子规则。同样的,我们现在使用 CI 而不是过渡态几何来进行量子力学推断。
具有 2 个或更多波相转变 (对称轴/多个异面)的系统:高阶 Möbius 芳香性,Lk=1。
2005 年,人们意识到具有两次波相转变的拓扑物也很有趣。虽然在很大程度上超出了启蒙的范畴,但值得注意的是,在环共轭 π 电子中的两次波相转变给出的 Möbius 芳香分子是符合 4n+2 规则的,但这一次该分子具有 C2 不对称手性,这不是与对称平面有关,而是与(三个或更多)轴有关。它的电子密度产生了一种称为环状连接的拓扑物,其环绕数 Lk=2。手性芳香族分子的化学(2005 年前被认为是矛盾的化合物)在迅速扩展。同样,我们可以用周环反应过渡态来类比。这将出现两个异面部分(异面的数量在形式上等于环绕数)。
基于 Fukui 前线轨道法的周环反应选律
这涉及使用量子力学原理为反应物生成最高占据分子轨道 (HOMO) 和最低未占据分子轨道 (LUMO)。新 σ 键的形成必须通过这些轨道节点的适当重叠来发生。如果只有一个 σ 键形成,如在电环化反应中,则仅考虑反应物的 HOMO 重叠即可,这种重叠可能以两种基本方式之一发生。同面模式涉及新 σ 键的每个组成部分,由反应物 π 系统的同一面生成。参见 DOI:10.1016/S0040-4039(01)83901-0。
现代方法,涉及势能面中过渡态和圆锥交错点的位置和分子动力学。
超越 FMO 方法的下一个层次的理论涉及:
- 热反应的过渡态
- 光化学反应的圆锥交错点
使用量子力学势能面和动力学明确周环反应的位置。
另,关于纽结理论和化学:
首次尝试从理论上推导元素周期表是在 1867 年,它是基于纽结,而不是当时还没有发明的量子力学。更令人毛骨悚然的是,纽结理论是弦理论的基础,弦理论试图解释所有基本粒子的内部结构。纽结理论相关可以看这个或这个
反应实例
电环化反应
1
最早在 1958 年,Vogel 报道了 (DOI:10.1002/jlac.19586150103) 环丁烯开环时意料之外的立体化学,如图。这是一个四电子体系。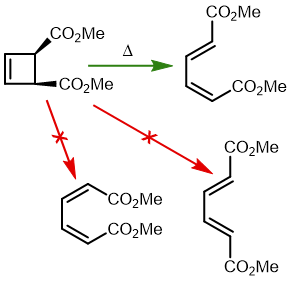
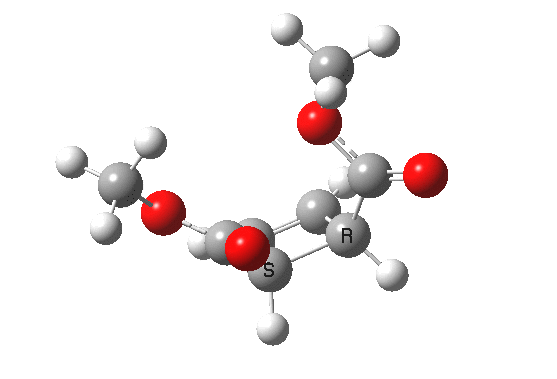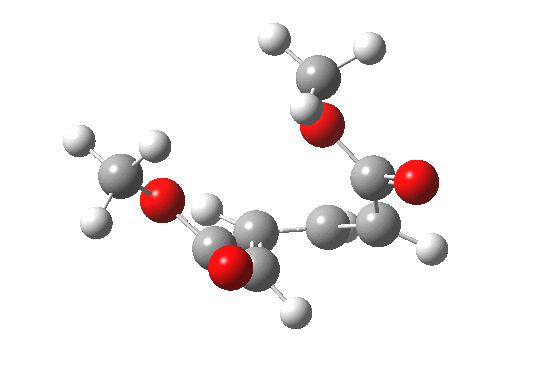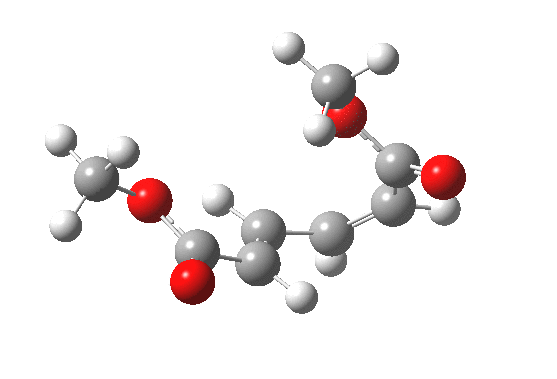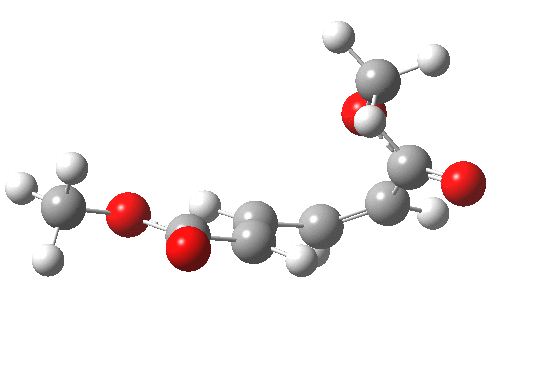
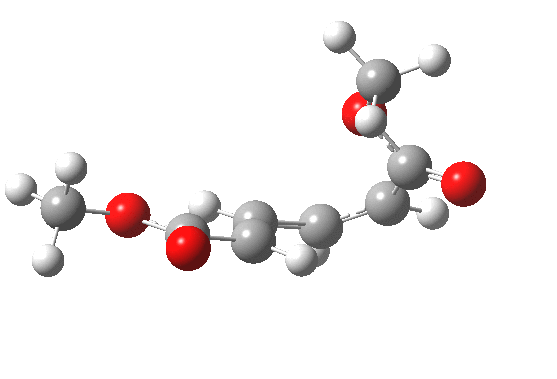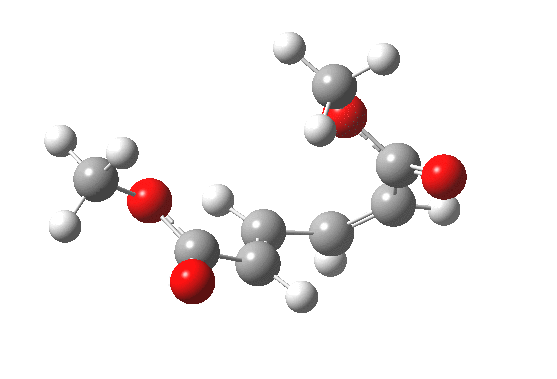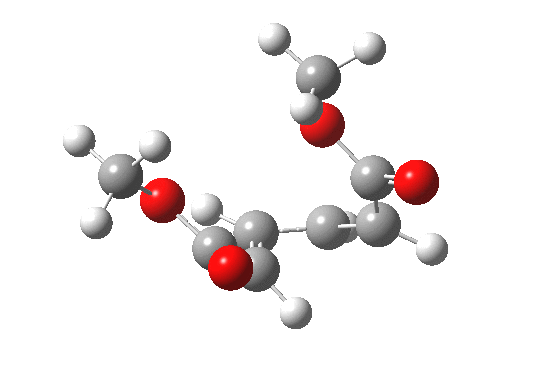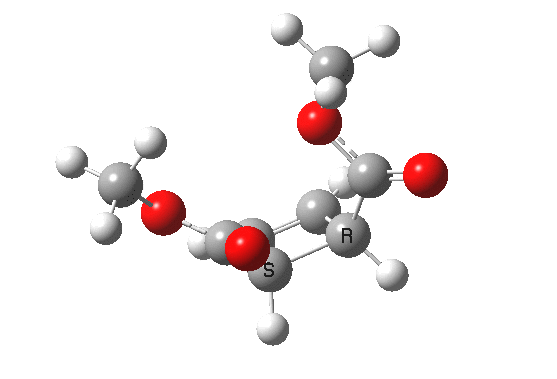
根据上节叙述的规则,加热时反应会以一个异面的 Möbius 芳香过渡态进行。两个烯基会向同向旋转 (顺旋 conrotation)。
为了形成新的 sigma 键,丁二烯片段的 HOMO 必须如图旋转,一个节点来自分子平面上方,另一个节点来自下方 (即异面)。Rzepa 制作过这个轨道的 3D 打印模型。
~~ 这都能出周边是吧 ~~
2
在光化学条件下,预计 4n 电子体系反应将通过同面且具有(假定的)对称面的 Hückel 芳香过渡态进行,例如:
从前线轨道(基态中的 LUMO,但处于被单个电子占据的激发态,因此实际上是 HOMO)可以再次看出这一点的含义。Rzepa 也制作过这个轨道的 3D 打印模型
一种更现代的理解光化学反应的方法是寻找圆锥交错点而非过渡态,详见下节。

下图计算结果表明反应具有同面的特征,但没有保留对称面。
3
环烯丙基阳离子的开环只涉及一对共轭电子,数量为 2 (4n+2,n=0)。在加热条件下,这将通过同面的 Hückel 芳香过渡态,两侧基团向相反方向向外旋转(对旋),并在此过程中保持对称面。
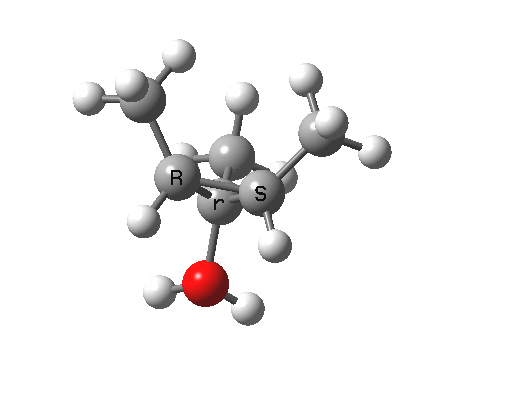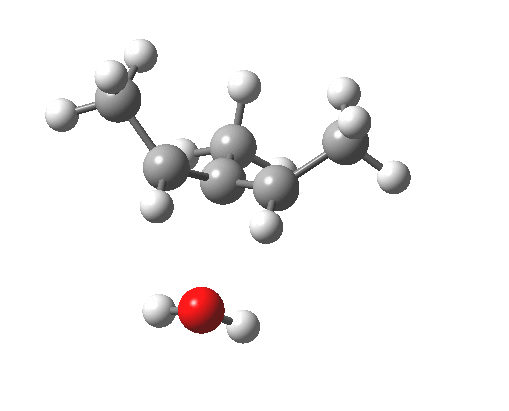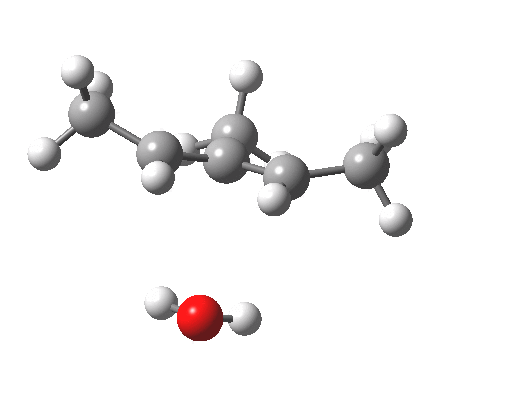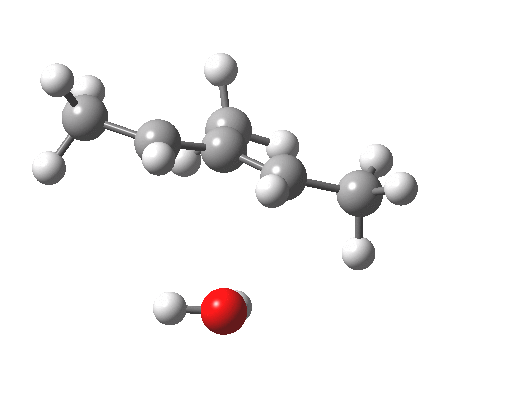
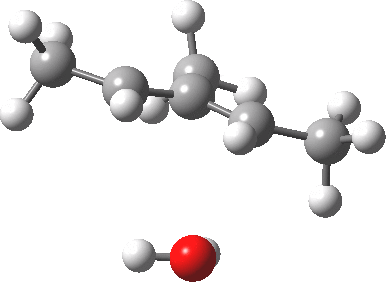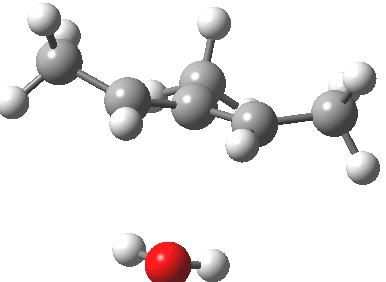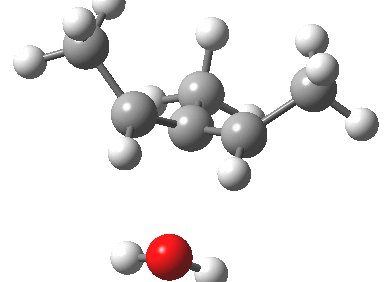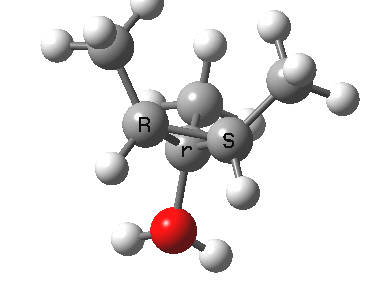
注意在过渡态中,离去基团的解离过程和电环化开环不是彼此独立的,这两者共同构成了一个单一的协同过程,形式为溶剂辅助的周环反应机理。
4
在涉及杂原子的情况下,孤立的原子对算作两个电子。因此,氮杂环丙烷的开环是一个具有一个反面的 4 电子过程 (=4N),通过 Möbius 芳香过渡态进行,两个甲基在相同的方向上旋转(顺旋),近似保持轴对称。
5
在 1961 年 Havinga 和 Schlatmann 对类固醇的研究中即观察到了六电子电环化的实例。1963 年,Corey 在二氢木香烃内酯的合成过程中得到了一个明确而清晰的周环反应立体选择性的例子,但都没有继续推进得出一个一般性的结论。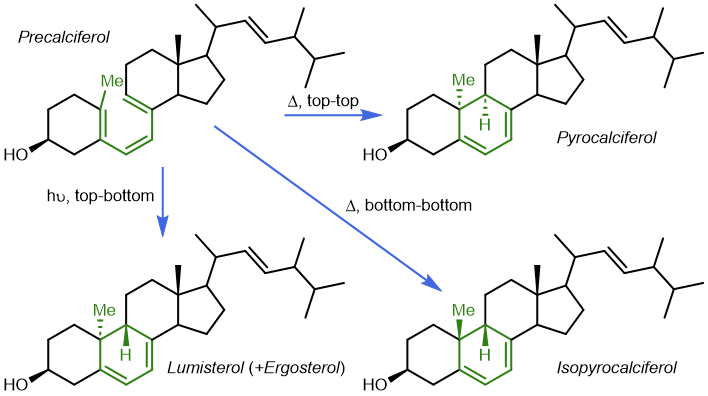

6
计算电子数时的一个重点是只包括环共轭体系。在这个例子中,对于[16]轮烯的一个价键异构体(具有 Cs 点群(即对称面),因此至少形式上是具有 4N 电子的 Hückel 反芳香性体系),其可发生两个独立的六电子 (4N+2) 电环化反应,并且中心双键中的电子不被计算在内,因为它们只是“旁观者”,因此 16 个电子中的 4 个不参与反应 (否则将是禁阻的 π4s+π4s 反应)。两个外环为 4N+2 Hückel 芳香族过渡态,中环为 4N 电子反芳香族旁观者。事实上,如果两个电环反应是分步进行的而非同时发生的,中心环上的 Hückel 反芳香性也可以避免,这就是实际发生的情况。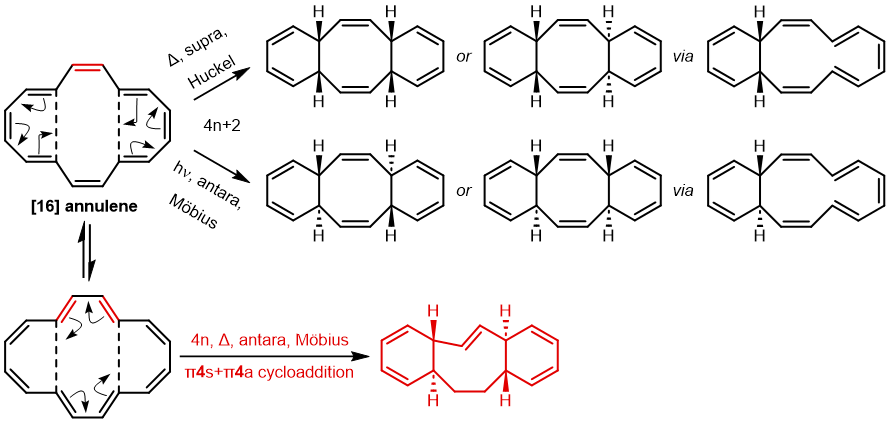
如果[16]环烯被绘制为不同的价键异构体,它可以拥有不同的构象,因为中心 C=C 键变为 C-C 键,其可以在室温下围绕该键发生旋转。这种新构象具有 C2 对称轴而非对称面,环化的过渡态具有 4n 电子,即 π4a+π4s 的 Möbius 芳香过渡态,中心有一个异面部分。这一次,二烯及其每侧的 8 个电子成为了“旁观者”(否则会构成两个禁阻的 4n+2 电子电环化过渡态)。反应中中心环为 4n 电子 Möbius 芳香过渡态,两个外环为 4n+2 电子 Möbius 反芳香过渡态。它比之前的 Hückel 模式过渡态能量高 14.8 kcal/mol,部分原因是该路径有两个反芳香环。
| 4n+2 Hückel: Plane of symmetry | 4n Möbius: Axis of symmetry |
|---|---|
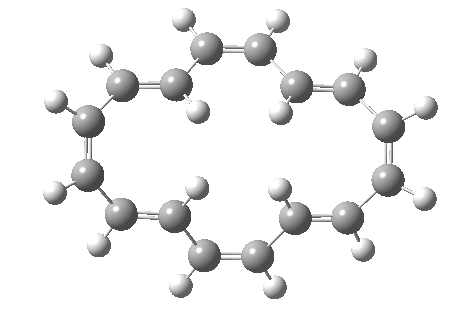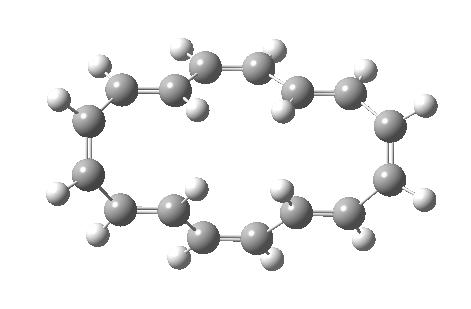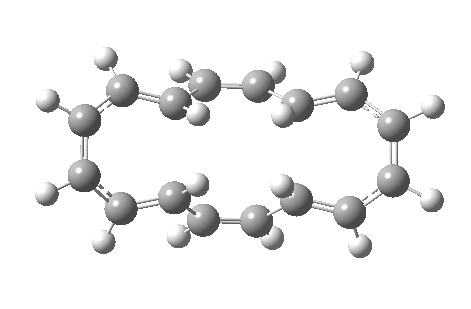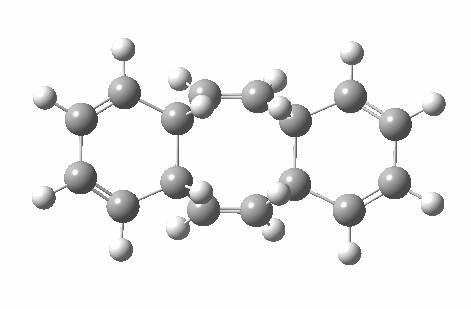 |
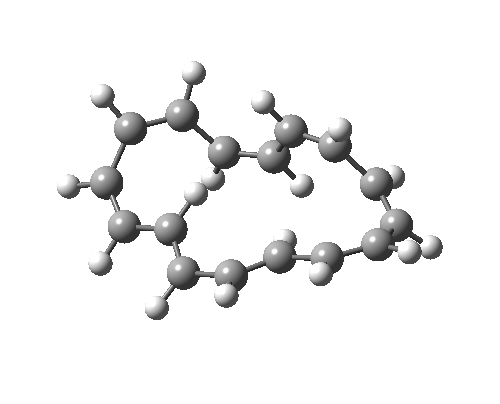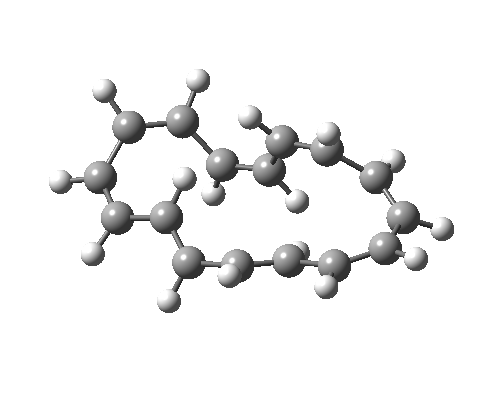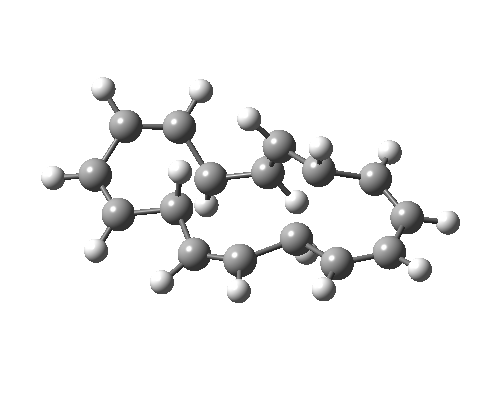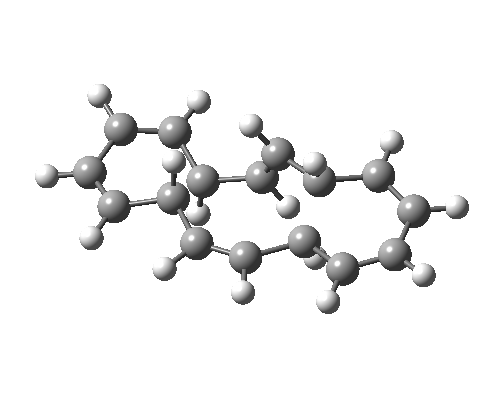 |
7
碳负离子提供 2e 的同面电环化
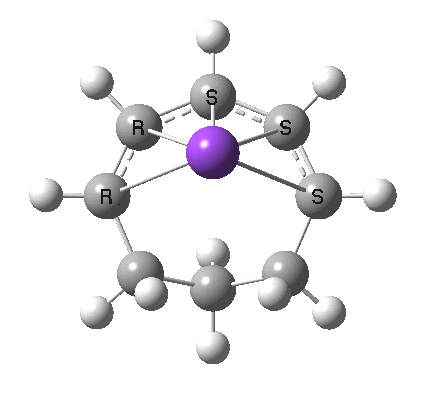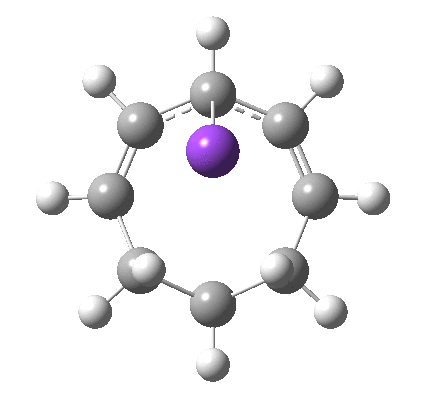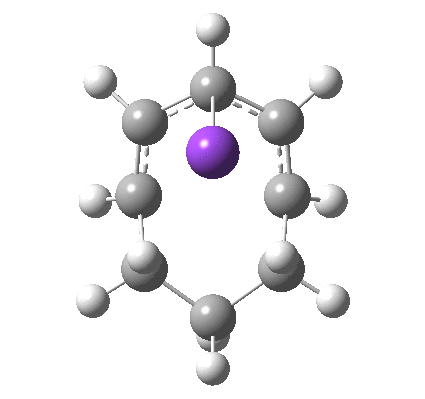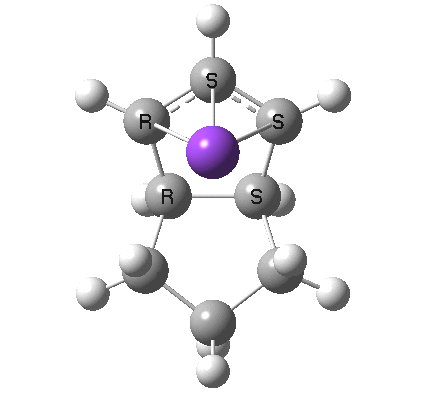
8
最后,想一想下面的电开环是通过 Hückel 还是 Möbius 过渡态发生的?
环加成反应